Recent Content

There is no such thing as a normal brain - TEDxPSU
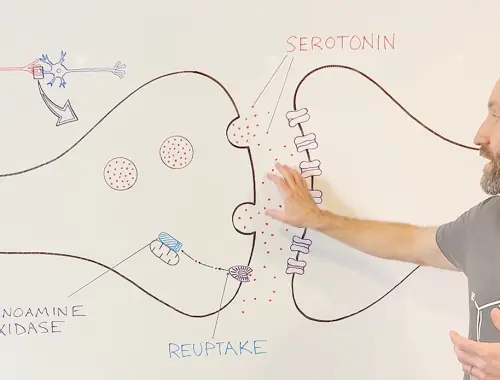
10-Minute Neuroscience: Depression

Strange Brains
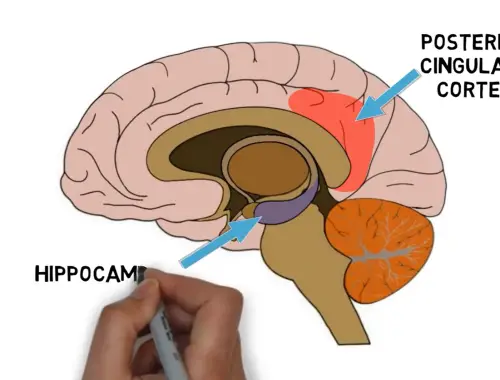
2-Minute Neuroscience: Sleepwalking
Glossary
Over 500 neuroscience terms defined, with supplemental imagery, videos, and links to related content.
Glossary

